Steps in Marple 
To go into details of the pipeline we dived the pipeline into modules similar to Hydra-Genetics module system. Default hydra-genetics settings/resources are used if no configuration is specified.
Prealignment
See the Prealignment hydra-genetics module documentation on ReadTheDoc or github documentation for more details on the softwares.

Pipeline output files
Only temporary intermediate files are created.
Trimming
Trimming of fastq files is performed by fastp v0.20.1.
Merging
Merging of fastq files belonging to the same sample are performed by simply concatenating the files with cat if applicable.
Alignment
See Alignment hydra-genetics module on ReadTheDocs or github for more information and documentation on the softwares used in the alignment steps.

Pipeline output files:
Results/{sample}/{sample}.cramResults/{sample}/{sample}.cram.crai
Alignment with BWA-mem
Alignment of fastq files into bam files is performed by bwa-mem v0.7.17 using the trimmed fastq files. This make it possible to speed up alignment by utilizing parallelization and also make it possible to analyze qc for lanes separately. Bamfiles are then directly sorted by samtools sort v1.15.
Read groups
Bam file read groups are set according to sequencing information in the units.tsv file.
The @RG read tag is set using the following options defined in the hydra-genetics bwa rule:
-R '@RG\tID:{ID}\tSM:{SM}\tPL:{PL}\tPU:{PU}\tLB:{LB}' -v 1
where the individual read groups are defined below:
| RG tag | Value |
|---|---|
| ID | sample_type.lane.barcode |
| SM | sample_type |
| PL | platform |
| PU | flowcell.lane.barcode |
| LB | sample_type |
Bam splitting
The bam files are split into chromosome files for faster performance in downstream analysis. Split files are used by markduplicates. Splitting is performed by samtools view v1.15.
Mark duplicates
Flagging duplicated reads are performed on individual chromosome bam files by picard MarkDuplicates v2.25.4.
Merging
Merging of deduplicated bam files belonging to the same sample are performed by samtools merge v1.15.
Sorting
Merged bamfile are sorted by samtools sort v1.15.
Bam indexing
Bamfile indexing is performed by samtools index v1.15.
SNV indels
SNV and indels are called using the SNV_indels (ReadTheDoc or github) module. Annotation is then done with Annotation module (ReadTheDocs or github).

Pipeline output files
Results/{sample}/{sample}.hard-filtered.vcf.gzResults/{sample}/{sample}.genome.vcf.gzResults/{sample}/mosaic/{sample}.deepmosaic.txtResults/{sample}/mosaic/{sample}.deepsomatic.vcf.gzResults/{sample}/mosaic/{sample}.mosaicforecast.phasingResults/{sample}/mosaic/{sample}.mosaicforecast.DEL.predictionsResults/{sample}/mosaic/{sample}.mosaicforecast.INS.predictionsResults/{sample}/mosaic/{sample}.mosaicforecast.SNP.predictions
SNV calling
Variants are called using DeepVariant. Deepvariant is run per chromosome over the regions defined in config["references"]["design_bed"]. The model type is set to "WES" and --output_gvcf is used to ensure that both genome vcf as well as a standard vcf is produced. The AF field is added to the INFO column in the vcf:s using the fix_af.py. The vcf header in the standard vcf is also updated to include a reference line using the add_ref_to_vcf.py to ensure that programs such as Alissa acknowledge the use of Hg38.
Normalizing
The standard vcf files is decomposed with vt decompose followed by vt decompose_blocksub v2015.11.10. The decomposed vcf files are then normalized by vt normalize v2015.11.10.
Annotation
The normalized standard VCF files are then annotated using VEP v109.3. Vep is run with the extra parameters --assembly GRCh38 --check_existing --pick --variant_class --everything.
See the annotation hydra-genetics module for additional information.
Mosaic variant calling
Possible mosaic variants is called by DeepSomatic which are then predicted to be real or not with MosaicForecast and DeepMosaic.
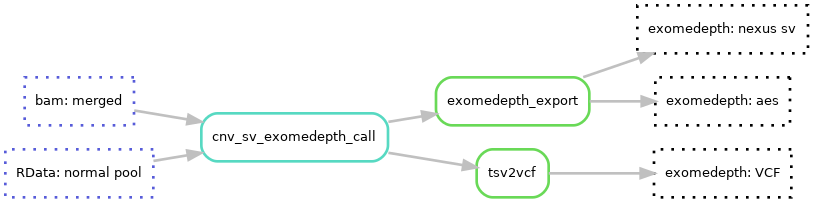
CNV
CNVs are called using the Hydra-Genetics CNV_SV module (ReadTheDocs or github).
Pipeline output files
Results/{sample}_{sequenceid}/{sample}_{sequenceid}_exomedepth_SV.txtResults/{sample}_{sequenceid}/{sample}_{sequenceid}_exomedepth.aedResults/{sample}/{sample}.cnv.vcf.gzResults/{sample}/mobile_elements/{sample}.ALU.vcf.gzResults/{sample}/mobile_elements/{sample}.LINE1.vcf.gzResults/{sample}/mobile_elements/{sample}.HERVK.vcf.gzResults/{sample}/mobile_elements/{sample}.SVA.vcf.gz
Exomedepth
To call larger structural variants Exomedepth v1.1.15 is used. Exomedepth does not use a window approach but evaluates each row in the bedfile as a segment, therefor the bedfile need to be split into appropriate large windows (e.g. using bedtools makewindows). Exomedepth also need a RData file containing the normal pool, this can be created using the Marple - references workflow. Lines with no-change calls (reads.ratio == 1) are removed from the output for Alissa compatibility. Since no sex-chromosome are included in the design Exomedepth is run with the same normalpool irregardless of the sample's biological sex. Marple is designed on HG38 therefor a genes and exonfile are also needed for annotation.
Mobile elements
To find mobile elements, like ALU, HERVK, LINE1 and SVA, MELT is used. It is free to use for academic purposes and you can buy a licence if used in other cases, like for this pipeline as part of the clinical workflow at the hospital. Since you should ask to use MELT, the singularity we use are local.
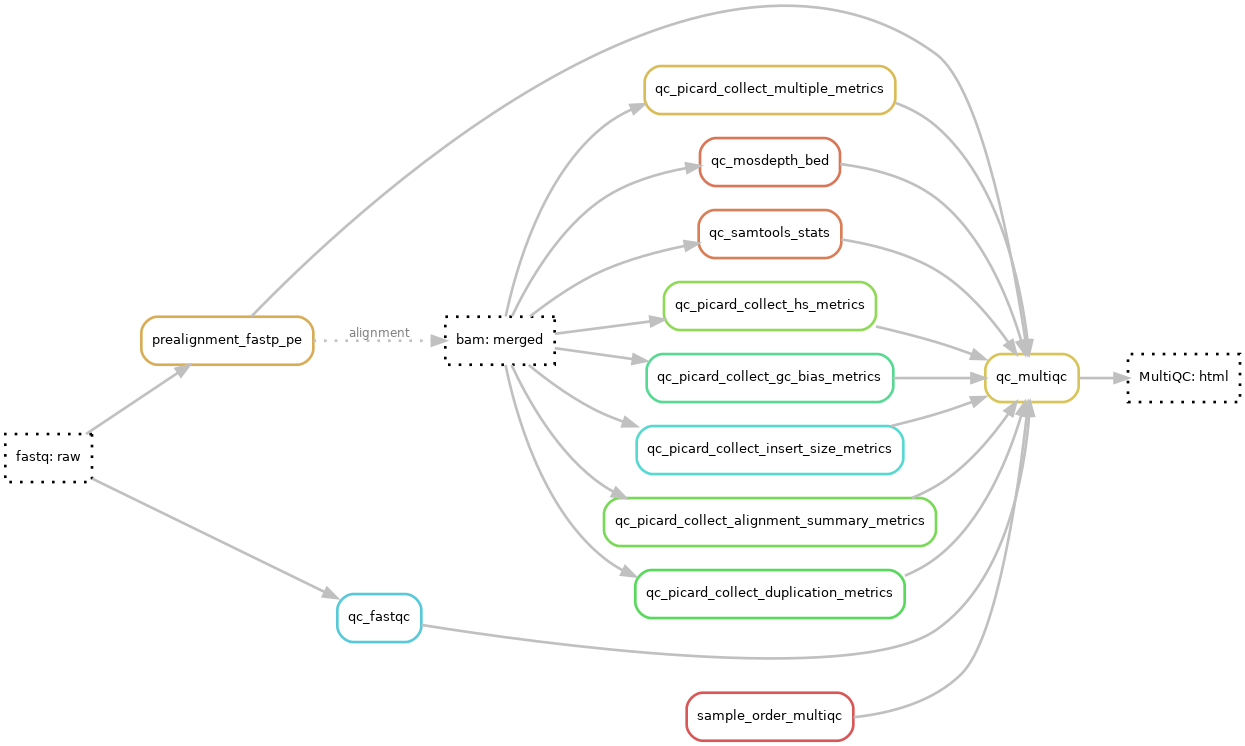
QC
For quality control the QC module (ReadTheDocs or github) is used and the results are summarized/aggregated into a MultiQC-report.
Pipeline output files
Results/{sequenceid}_MultiQC.html
MultiQC
A MultiQC html report is generated using MultiQC v1.11. The report starts with a general statistics table showing the most important QC-values followed by additional QC data and diagrams. See result files page for more in-depth info on general stats columns. The qc data is collected and generated using Fastp, FastQC, samtools, picard and Mosdepth.
The samples in the MultiQC report is renamed and ordered based on the order of the SampleSheet.csv to enable easier integration with the wet lab follow-up.
The report is configured based on a MultiQC config file.
Expand to view current MultiQC config.yaml
title: "Clinical Genomics MultiQC Report"
subtitle: "Twist Cancer"
intro_text: "The MultiQC report summarize analysis results from Twist Cancer data that been analyzed by the pipeline marple_rd_tc (https://github.com/clinical-genomics-uppsala/marple-rd-tc). Reference used: GRCh38."
report_header_info:
- Contact E-mail: "igp-klinsek-bioinfo@lists.uu.se"
- Application Type: "Twist Cancer Panel"
- Project Type: "Clinical Samples"
# - Sequencing Platform: "HiSeq 2500 High Output V4"
# - Sequencing Setup: "2x150"
decimalPoint_format: ','
## maste anpassa configen lite mera. 20x breadth, insert size och bases on target. Fold80?
extra_fn_clean_exts: ##from this until end
- '.duplication_metrics'
- '_N'
custom content:
order:
- fastqc
- mosdepth
- fastp
- peddy
- samtools
- picard
mosdepth_config:
include_contigs:
- "chr1"
- "chr2"
- "chr3"
- "chr4"
- "chr5"
- "chr6"
- "chr7"
- "chr8"
- "chr9"
- "chr10"
- "chr11"
- "chr12"
- "chr13"
- "chr14"
- "chr15"
- "chr16"
- "chr17"
- "chr18"
- "chr19"
- "chr20"
- "chr21"
- "chr22"
read_count_multiplier: 0.001
read_count_prefix: "K"
read_count_desc: "thousands"
table_columns_visible:
FastQC:
percent_duplicates: False
percent_gc: False
avg_sequence_length: False
percent_fails: False
total_sequences: False
fastp:
pct_adapter: True
pct_surviving: False
after_filtering_gc_content: False
filtering_result_passed_filter_reads: False
after_filtering_q30_bases: False
after_filtering_q30_rate: False
pct_duplication: False
mosdepth:
median_coverage: True
mean_coverage: False
1_x_pc: False
5_x_pc: False
10_x_pc: False
20_x_pc: False
30_x_pc: True
50_x_pc: True
Picard:
PCT_PF_READS_ALIGNED: False
summed_median: False
summed_mean: True
PERCENT_DUPLICATION: True
MEDIAN_COVERAGE: False
MEAN_COVERAGE: False
SD_COVERAGE: False
PCT_30X: False
PCT_TARGET_BASES_30X: False
FOLD_ENRICHMENT: False
TOTAL_READS: False
Samtools:
error_rate: False
non-primary_alignments: False
reads_mapped: False
reads_mapped_percent: True
reads_properly_paired_percent: True
reads_MQ0_percent: False
raw_total_sequences: True #only on bedfile not total of fastq, bases on target only
# Patriks plug in, addera egna columner till general stats
multiqc_cgs:
Picard:
FOLD_80_BASE_PENALTY:
title: "Fold80"
description: "Fold80 penalty from picard hs metrics"
min: 1
max: 3
scale: "RdYlGn-rev"
format: "{:.1f}"
PCT_SELECTED_BASES:
title: "Bases on Target"
description: "On+Near Bait Bases / PF Bases Aligned from Picard HsMetrics"
format: "{:.2%}"
ZERO_CVG_TARGETS_PCT:
title: "Target bases with zero coverage [%]"
description: "Target bases with zero coverage [%] from Picard"
min: 0
max: 100
scale: "RdYlGn-rev"
format: "{:.2%}"
Samtools:
average_quality:
title: "Average Quality"
description: "Ratio between the sum of base qualities and total length from Samtools stats"
min: 0
max: 60
scale: "RdYlGn"
mosdepth:
20_x_pc: #Cant get it to work
title: "20x percent"
description: "Fraction of genome with at least 20X coverage"
max: 100
min: 0
suffix: "%"
scale: "RdYlGn"
# Galler alla kolumner oberoende pa module!
table_columns_placement:
mosdepth:
median_coverage: 601
1_x_pc: 666
5_x_pc: 666
10_x_pc: 602
20_x_pc: 603
30_x_pc: 604
50_x_pc: 605
Samtools:
raw_total_sequences: 500
reads_mapped: 501
reads_mapped_percent: 502
reads_properly_paired_percent: 503
average_quality: 504
error_rate: 555
reads_MQ0_percent: 555
non-primary_alignments: 555
Picard:
TOTAL_READS: 500
PCT_SELECTED_BASES: 801
FOLD_80_BASE_PENALTY: 802
PCT_PF_READS_ALIGNED: 888
summed_median: 888
PERCENT_DUPLICATION: 803
summed_mean: 804
STANDARD_DEVIATION: 805
ZERO_CVG_TARGETS_PCT: 888
MEDIAN_COVERAGE: 888
MEAN_COVERAGE: 888
SD_COVERAGE: 888
PCT_30X: 888
PCT_TARGET_BASES_30X: 888
FOLD_ENRICHMENT: 888
FastQC
FastQC v0.11.9 is run on the raw fastq-files before trimming.
Mosdepth
Mosdepth v0.3.2 is used together with a bedfile covering all coding exons (config[reference][exon_bed]) and thresholds (10,20,50) to calculate coverage.
Samtools
Samtools stats v1.15 is run on BWA-mem aligned and merged bam files without any designfile.
Picard
Picard v2.25.4 is run on BWA-mem aligned and merged bam files collecting a number of metrics. The metrics calculated are listed below:
- picard CollectAlignmentSummaryMetrics - using fasta reference genome file
- picard CollectDuplicationMetrics
- picard CollectGCBiasMetrics
- picard CollectHsMetrics - using fasta reference genome file, the full capture design bedfile (
config[reference][design_bed]), and with the option COVERAGE_CAP=5000 - picard CollectInsertSizeMetrics
- picard CollectMultipleMetrics - using fasta reference genome file, and the full bedfile (
config[reference][design_intervals])